
FDA approves eflornithine for adult and pediatric patients with high-risk neuroblastoma
FDA approves eflornithine for adult and pediatric patients with high-risk neuroblastoma
First FDA approval of a therapy intended to reduce the risk of relapse in pediatric patients with HRNB
Efficacy was evaluated in an externally controlled trial comparing outcomes from Study 3b (investigational arm) and Study ANBL0032 (clinical trial-derived external control arm)
EFS HR was 0.48 (95% CI: 0.27, 0.85) and OS HR was 0.32 (95% CI: 0.15, 0.70)

On December 13, 2023, the Food and Drug Administration approved eflornithine (IWILFIN, USWM, LLC) to reduce the risk of relapse in adult and pediatric patients with high-risk neuroblastoma (HRNB) who have demonstrated at least a partial response to prior multiagent, multimodality therapy including anti-GD2 immunotherapy.
This represents the first FDA approval of a therapy intended to reduce the risk of relapse in pediatric patients with HRNB.
Full prescribing information for IWILFIN will be posted here.
Efficacy was evaluated in an externally controlled trial comparing outcomes from Study 3b (investigational arm) and Study ANBL0032 (clinical trial-derived external control arm). Study 3b (NCT02395666) was a multi-center, open label, non-randomized trial with two cohorts. A total of 105 eligible patients with HRNB from one cohort (Stratum 1) received eflornithine orally, twice daily at a dosage based on body surface area (BSA) until disease progression, unacceptable toxicity, or for a maximum of 2 years. Study 3b was prospectively designed to compare outcomes to the historical benchmark event free survival (EFS) rate from Study ANBL0032 reported in published literature.
The external control arm was derived from 1,241 patients on the experimental arm of Study ANBL0032, a multi-center, open-label, randomized trial of dinutuximab, granulocyte-macrophage colony-stimulating factor, interleukin-2, and cis-retinoic acid compared to cis-retinoic acid alone in pediatric patients with HRNB.
Patients who met the criteria for the comparative analysis of Study 3b and ANBL0032, with complete data for specified clinical covariates, were matched (1:3) using propensity scores; the matched efficacy populations for the primary analysis included 90 patients treated with IWILFIN and 270 control patients from Study ANBL0032.
The major efficacy outcome measure was event free survival (EFS), defined as disease progression, relapse, secondary cancer, or death due to any cause. An additional efficacy outcome measure was overall survival (OS), defined as death due to any cause. In the protocol-specified primary analysis, the EFS hazard ratio (HR) was 0.48 (95% CI: 0.27, 0.85) and OS HR was 0.32 (95% CI: 0.15, 0.70). Given the uncertainty in treatment effect estimation associated with the externally controlled study design, supplementary analyses in subpopulations or using alternative statistical methods were performed. In these analyses, the EFS HR ranged from 0.43 (95% CI: 0.23, 0.79) to 0.59 (95% CI: 0.28, 1.27), and the OS HR ranged from 0.29 (95% CI: 0.11, 0.72) to 0.45 (95% CI: 0.21, 0.98).
The most common adverse reactions (≥5%) in Study 3b, including laboratory abnormalities, were otitis media, diarrhea, cough, sinusitis, pneumonia, upper respiratory tract infection, conjunctivitis, vomiting, pyrexia, allergic rhinitis, decreased neutrophils, increased ALT, increased AST, hearing loss, skin infection, and urinary tract infection.
The recommended dose is based on BSA. See the prescribing information for more information.
This review used the Real-Time Oncology Review (RTOR) pilot program, which streamlined data submission prior to the filing of the entire clinical application, and the Assessment Aid, a voluntary submission from the applicant to facilitate the FDA’s assessment.
This application was granted priority review, breakthrough designation, and orphan drug designation. FDA expedited programs are described in the Guidance for Industry: Expedited Programs for Serious Conditions-Drugs and Biologics.


European Commission Grant Conditional MA to Pfizer’s ELREXFIO® for Relapsed and Refractory Multiple Myeloma
European Commission Grant Conditional MA to Pfizer’s ELREXFIO® for Relapsed and Refractory Multiple Myeloma
CMA based on clinically meaningful response rates, duration of response, and safety from the Phase 2 MagnetisMM-3 trial
ORR 61.0% (75/123); 35.0% ≥complete response. Fifty responders switched to biweekly dosing, and 40 (80.0%) improved or maintained their response for ≥6 months
With median follow-up of 14.7 months, median duration of response, PFS & OS (secondary endpoints) have not been reached

Conditional marketing authorization is based on clinically meaningful response rates, duration of response, and safety from the Phase 2 MagnetisMM-3 trial
ELREXFIO is an off-the-shelf (ready-to-use), fixed-dose, subcutaneous BCMA-directed bispecific antibody immunotherapy with reduced dosing after 24 weeks for responding patients
NEW YORK--(BUSINESS WIRE)-- Pfizer Inc. (NYSE:PFE) today announced the European Commission (EC) has granted conditional marketing authorization for ELREXFIO® (elranatamab). ELREFXIO is a targeted immunotherapy for the treatment of adult patients with relapsed and refractory multiple myeloma (RRMM) who have received at least three prior therapies, including a proteasome inhibitor, an immunomodulatory agent, and an anti-CD38 antibody and have demonstrated disease progression on the last therapy. ELREXFIO is an off-the-shelf (ready-to-use) B-cell maturation antigen (BCMA)-CD3-directed bispecific antibody (BsAb) immunotherapy that induces deep and durable responses, with a manageable tolerability profile as well as convenient subcutaneous dosing.
"More than 50,000 Europeans are diagnosed with multiple myeloma each year, and too often, they face relapse and treatment resistance,” said Chris Boshoff, Chief Oncology Research and Development Officer and Executive Vice President, Pfizer. “Today's approval provides a new, broadly available option for people with hard-to-treat multiple myeloma, and we continue to explore the use of ELREXFIO in earlier lines of treatment so that more people may ultimately benefit from this therapy."
The conditional marketing authorization for ELREXFIO is valid in all 27 EU member states as well as Iceland, Liechtenstein, and Norway. This authorization follows the European Medicines Agency (EMA) Committee for Medicinal Products for Human Use (CHMP) recommendation for a conditional marketing authorization on October 12, 2023.
Authorization was based on data from cohort A of the Phase 2 MagnetisMM-3 study (NCT04649359) showing meaningful responses among heavily pretreated RRMM patients – at least three prior therapies, including an immunomodulatory agent, a proteasome inhibitor, and an anti-CD38 antibody – who received ELREXFIO as their first BCMA-directed therapy. In an analysis of these patients (n=123), the objective response rate was 61%, with a 71% probability of maintaining a response at 15 months.
The results from MagnetisMM-3 also established once-every-other-week dosing with ELREXFIO for all responding patients after 24 weeks of weekly therapy, which means less time at the clinic and potentially greater long-term treatment tolerability. Among responding patients who switched to every-other-week dosing at least six months prior to the data cut-off date (n=50), 80% maintained or improved their response after the switch, with 38% attaining a complete response (CR) or better after the switch. These data were published in Nature Medicine.
The most common adverse reactions to ELREXFIO are cytokine release syndrome (CRS) (58%), anemia (54%), neutropenia (45%), fatigue (44%), upper respiratory tract infection (39%), injection site reaction (38%), diarrhea (38%), pneumonia (37%), thrombocytopenia (36%), lymphopenia (30%), decreased appetite (27%), rash (26%), joint pain (arthralgia) (25%), fever (pyrexia) (27%), hypokalemia (23%), nausea (21%), and dry skin (21%). Serious adverse reactions are pneumonia (31%), sepsis (15%), CRS (13%), anemia (6%), upper respiratory tract infection (5%), urinary tract infection (3%), febrile neutropenia (3%), dyspnea (2%), and pyrexia (2%). Most cases of CRS were Grade 1 (44% of patients), with Grade 2 in 14% and Grade 3 in less than 1% of patients.
Due to the risk of CRS and immune effector cell-associated neurotoxicity syndrome (ICANS), patients should be monitored for signs and symptoms for 48 hours after administration of each of the two step-up doses within the ELREXFIO dosing schedule and instructed to remain in proximity of a healthcare facility. In the EU, precautionary hospitalization is not required. Patients are not required to stay near a healthcare facility for the 76-mg first treatment dose.
About Multiple Myeloma
Multiple myeloma (MM) is an aggressive and currently incurable blood cancer that affects plasma cells made in the bone marrow. Healthy plasma cells make antibodies that help the body fight infection.1 MM is the second most common type of blood cancer, with over 50,000 new cases diagnosed annually in Europe and 176,000 new cases diagnosed globally each year.2,3 About 40% of those diagnosed with MM won’t survive beyond five years,4 and most will receive four or more lines of therapy due to relapse.5 While disease trajectory varies for each person, relapses are nearly inevitable.6 The goal of therapy for people with RRMM is to achieve disease control with acceptable toxicity and improved quality of life.7
About ELREXFIO® (elranatamab)
ELREXFIO is a subcutaneously delivered B-cell maturation antigen (BCMA)-CD3-directed bispecific antibody (BsAb) immunotherapy that binds to BCMA on myeloma cells and CD3 on T-cells, activating the T-cells to kill myeloma cells.
In August 2023, ELREXFIO was approved by the U.S. Food and Drug Administration (FDA) under its Accelerated Approval Program, which allows for earlier approval of drugs that treat serious conditions and fill an unmet medical need. ELREXFIO has also received approval in Switzerland and Brazil under Project Orbis, a framework for the concurrent submission and review of oncology drugs among international partners to potentially expedite approvals. Three other countries (Canada, Australia, and Singapore) are participating in Project Orbis. The UK Medicines and Healthcare Products Regulatory Agency (MHRA) has granted ELREXFIO Innovative Medicine Designation and the Innovation Passport for the treatment of MM.
Pfizer’s extensive MagnetisMM clinical development program is continuing to investigate ELREXFIO’s use across the entire spectrum of patients with MM, from RRMM to newly diagnosed MM. Ongoing registrational-intent trials are exploring ELREXFIO both as monotherapy and in combination with standard or novel therapies. These include MagnetisMM-5 in the double-class exposed setting, MagnetisMM-6 in newly diagnosed patients who are ineligible for stem cell transplant, and MagnetisMM-7 in newly diagnosed patients after transplant.
Detailed information on this medicinal product is available on the website of the European Medicines Agency http://www.ema.europa.eu.
U.S. INDICATION
ELREXFIO may cause side effects that are serious, life-threatening, or can lead to death, including cytokine release syndrome (CRS) and neurologic problems. CRS is common during treatment with ELREXFIO.
Tell your healthcare provider or get medical help right away if you develop any signs or symptoms of CRS or neurologic problems, including:
fever of 100.4°F (38°C) or higher
trouble breathing
chills
dizziness or light-headedness
fast heartbeat
headache
increased liver enzymes in your blood
agitation, trouble staying awake, confusion or disorientation, or seeing or hearing things that are not real (hallucinations)
trouble speaking, thinking, remembering things, paying attention, or understanding things
problems walking, muscle weakness, shaking (tremors), loss of balance, or muscle spasms
numbness and tingling (feeling like “pins and needles”)
burning, throbbing, or stabbing pain
changes in your handwriting
Due to the risk of CRS, you will receive ELREXFIO on a “step-up” dosing schedule and should be hospitalized for 48 hours after the first “step-up” dose and for 24 hours after the second “step-up” dose of ELREXFIO.
For your first dose, you will receive a smaller “step-up” dose of ELREXFIO on day 1
For your second dose, you will receive a larger “step-up” dose of ELREXFIO, which is usually given on day 4 of treatment
For your third dose, you will receive the first “treatment” dose of ELREXFIO, which is usually given on day 8
If your dose of ELREXFIO is delayed for any reason, you may need to repeat step-up dosing. Before each dose of ELREXFIO you receive during the step-up dosing schedule, you will receive medicines to help reduce your risk of CRS. Your healthcare provider will decide if you need to receive medicines to help reduce your risk of CRS with future doses.
ELREXFIO is available only through the ELREXFIO Risk Evaluation and Mitigation Strategy (REMS) Program due to the risk of CRS and neurologic problems. You will receive an ELREXFIO Patient Wallet Card from your healthcare provider. Carry the ELREXFIO Patient Wallet Card with you at all times and show it to all of your healthcare providers. The ELREXFIO Patient Wallet Card lists signs and symptoms of CRS and neurologic problems. Get medical help right away if you develop any of the signs and symptoms listed on the ELREXFIO Patient Wallet Card. You may need to be treated in a hospital.
Before taking ELREXFIO, tell your healthcare provider about all of your medical conditions, including if you:
have an infection
are pregnant or plan to become pregnant. ELREXFIO may harm your unborn baby. Females who are able to become pregnant should do a pregnancy test before starting treatment with ELREXFIO and should use effective birth control during treatment and for 4 months after your last dose of ELREXFIO. Tell your healthcare provider right away if you become pregnant or think that you may be pregnant during treatment with ELREXFIO
are breastfeeding or plan to breastfeed. It is not known if ELREXFIO passes into your breast milk. Do not breastfeed during treatment and for 4 months after your last dose of ELREXFIO
Tell your healthcare provider about all of the medications you take, includingprescription and over-the-counter medications, vitamins, and herbal supplements.
Do not drive, operate heavy or potentially dangerous machinery, or do other dangerous activities during treatment with ELREXFIO:
for 48 hours after completing each of the 2 doses of ELREXFIO that are part of the “step-up dosing schedule” and your first full treatment dose, and
at any time during treatment with ELREXFIO if you develop any new neurologic symptoms, such as dizziness, confusion, shaking (tremors), sleepiness, or any other symptom that impairs consciousness, until the symptoms go away.
Infections: Upper respiratory tract infection and pneumonia are common during treatment with ELREXFIO. ELREXFIO can cause bacterial and viral infections that are severe, life-threatening, or that may lead to death.
Your healthcare provider may prescribe medications for you to help prevent infections and treat you as needed if you develop an infection during treatment with ELREXFIO
Tell your healthcare provider right away if you develop any signs or symptoms of an infection during treatment with ELREXFIO, including: fever of 100.4°F (38°C) or higher, chills, cough, shortness of breath, chest pain, sore throat, pain during urination, or feeling weak or generally unwell
People with active infections should not start ELREXFIO
Decreased white blood cell counts: Decreased white blood cell counts are common during treatment with ELREXFIO and can also be severe. A fever can occur with low white blood cell counts and may be a sign that you have an infection. Your healthcare provider will treat you as needed.
Liver problems: ELREXFIO can cause increased liver enzymes and bilirubin in your blood. These increases can happen with or without you also having CRS. Tell your healthcare provider if you develop any of the following signs or symptoms of a liver problem, including:
tiredness
loss of appetite
pain in your right upper stomach-area
dark urine
yellowing of your skin or the white part of your eyes
The most common side effects of ELREXFIO include:
tiredness
injection site reaction, such as redness, itching, pain, bruising, rash, swelling, and tenderness
diarrhea
muscle and bone pain
decreased appetite
rash
cough
nausea
fever
The most common severe abnormal lab test results with ELREXFIO include decreased white blood cells, red blood cells, and platelets.
Your healthcare provider may temporarily or permanently stop ELREXFIO if you have any of the side effects listed and they are severe. These are not all of the possible side effects of ELREXFIO.
Call your healthcare provider for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
What is ELREXFIO?
ELREXFIO is a prescription medication used to treat adults with multiple myeloma who:
have already received at least 4 treatment regimens, including a proteasome inhibitor, an immunomodulatory agent, and an anti-CD38 monoclonal antibody, to treat their multiple myeloma, and
their cancer has come back or did not respond to prior treatment.
ELREXFIO was approved based on patient responses and durability of response. There are ongoing studies to confirm its clinical benefit. It is not known if ELREXFIO is safe and effective in children.
Please read full Prescribing Information, including BOXED WARNING, for ELREXFIO.
About Pfizer Oncology
At Pfizer Oncology, we are committed to advancing medicines wherever we believe we can make a meaningful difference in the lives of people living with cancer. Today, we have an industry-leading portfolio of 24 approved innovative cancer medicines and biosimilars across more than 30 indications, including breast, genitourinary, colorectal, blood, and lung cancers, as well as melanoma.
About Pfizer: Breakthroughs That Change Patients’ Lives
At Pfizer, we apply science and our global resources to bring therapies to people that extend and significantly improve their lives. We strive to set the standard for quality, safety and value in the discovery, development and manufacture of health care products, including innovative medicines and vaccines. Every day, Pfizer colleagues work across developed and emerging markets to advance wellness, prevention, treatments and cures that challenge the most feared diseases of our time. Consistent with our responsibility as one of the world’s premier innovative biopharmaceutical companies, we collaborate with health care providers, governments and local communities to support and expand access to reliable, affordable health care around the world. For more than 170 years, we have worked to make a difference for all who rely on us. We routinely post information that may be important to investors on our website at www.Pfizer.com. In addition, to learn more, please visit us on www.Pfizer.com and follow us on Twitter at @Pfizer and @Pfizer News, LinkedIn, YouTube and like us on Facebook at Facebook.com/Pfizer.
Disclosure Notice
The information contained in this release is as of December 8, 2023. Pfizer assumes no obligation to update forward-looking statements contained in this release as the result of new information or future events or developments.
This release contains forward-looking information about ELREXFIO (elranatamab), a B-cell maturation antigen (BCMA) CD3-directed bispecific antibody, including its potential benefits, an approval by the European Commission for the treatment of adult patients with relapsed and refractory multiple myeloma (RRMM) who have received at least three prior therapies, including an immunomodulatory agent, a proteasome inhibitor and an anti-CD38 antibody and have demonstrated disease progression on the last therapy, pending regulatory applications and the MagnetisMM clinical program to potentially expand ELREXFIO into earlier lines of treatment, as monotherapy and in combination with standard or novel therapies, and Pfizer Oncology, that involves substantial risks and uncertainties that could cause actual results to differ materially from those expressed or implied by such statements. Risks and uncertainties include, among other things, uncertainties regarding the commercial success of ELREXFIO; the uncertainties inherent in research and development, including the ability to meet anticipated clinical endpoints, commencement and/or completion dates for our clinical trials, regulatory submission dates, regulatory approval dates and/or launch dates, as well as the possibility of unfavorable new clinical data and further analyses of existing clinical data; the risk that clinical trial data are subject to differing interpretations and assessments by regulatory authorities; whether regulatory authorities will be satisfied with the design of and results from our clinical studies; whether and when drug applications for any potential indications for ELREXFIO may be filed in any particular jurisdictions; whether and when regulatory authorities in any jurisdictions may approve any applications that may be pending or filed for ELREXFIO, which will depend on myriad factors, including making a determination as to whether the product's benefits outweigh its known risks and determination of the product's efficacy and, if approved, whether ELREXFIO will be commercially successful; decisions by regulatory authorities impacting labeling, manufacturing processes, safety and/or other matters that could affect the availability or commercial potential of ELREXFIO; uncertainties regarding the impact of COVID-19 on Pfizer’s business, operations and financial results; and competitive developments.
A further description of risks and uncertainties can be found in Pfizer’s Annual Report on Form 10-K for the fiscal year ended December 31, 2022 and in its subsequent reports on Form 10-Q, including in the sections thereof captioned “Risk Factors” and “Forward-Looking Information and Factors That May Affect Future Results,” as well as in its subsequent reports on Form 8-K, all of which are filed with the U.S. Securities and Exchange Commission and available at www.sec.gov and www.pfizer.com.
1 Multiple Myeloma Research Foundation (MMRF): What is Multiple Myeloma?; Accessed August 22, 2023; Available at: https://themmrf.org/multiple-myeloma
2 Myeloma Patients Europe: Myeloma A Patients Guide; Updated May 2022. Accessed August 2, 2023. Available at: https://www.mpeurope.org/wp-content/uploads/2023/01/Myeloma-Patients-Guide.pdf.
3 World Health Organization: Globocan 2020: Multiple Myeloma; Accessed August 22, 2023. Available at: https://gco.iarc.fr/today/data/factsheets/cancers/35-Multiple-myeloma-fact-sheet.pdf
4 National Cancer Institute. Surveillance, Epidemiology, and End Results Program. Cancer Stat Facts: Myeloma. Accessed on Oct. 30, 2023. Available at: https://seer.cancer.gov/statfacts/html/mulmy.html
5 Mikhael, J, Ismaila N, Cheung M, et al. Treatment of multiple myeloma: ASCO and CCO joint clinical practice guideline. J Clin Oncol. 37:1228-1263.
6 Dimopoulos MA, Richardson P, Lonial S. Treatment options for patients with heavily pretreated relapsed and refractory multiple myeloma. Clin Lymphoma Myeloma Leuk. 2022;22(7):460-473. doi:10.1016/j.clml.2022.01.011
7 Bazarbachi AH, Al Hamed R, Malard F, Harousseau JL, Mohty M. Relapsed refractory multiple myeloma: a comprehensive overview. Leukemia. 2019 Oct;33(10):2343-57.


FDA grants accelerated approval to pirtobrutinib for 3L CLL/SLL
FDA grants accelerated approval to pirtobrutinib for 3L CLL/SLL
Approval based on BRUIN (NCT03740529], an open-label, single-arm, multicohort trial (N=108)
ORR was 72% (95% CI: 63, 80), DOR was 12.2 months (95% CI: 9.3, 14.7). All responses were partial responses

On December 1, 2023, the Food and Drug Administration granted accelerated approval to pirtobrutinib (Jaypirca, Eli Lilly and Company) for adults with chronic lymphocytic leukemia or small lymphocytic lymphoma (CLL/SLL) who have received at least two prior lines of therapy, including a BTK inhibitor and a BCL-2 inhibitor.
Prescribing information for Jaypirca will be posted here.
Efficacy was evaluated in BRUIN (NCT03740529], an open-label, international, single-arm, multicohort trial that included 108 patients with CLL or SLL previously treated with at least two prior lines of therapy, including a BTK inhibitor and a BCL-2 inhibitor. Patients received a median of 5 prior lines of therapy (range: 2 to 11). Seventy-seven percent of patients discontinued the last BTK inhibitor for refractory or progressive disease. Pirtobrutinib was administered orally at 200 mg once daily and was continued until disease progression or unacceptable toxicity.
The main efficacy outcome measures were overall response rate (ORR) and duration of response (DOR), as assessed by an independent review committee using 2018 iwCLL criteria. The ORR was 72% (95% CI: 63, 80) and median DOR was 12.2 months (95% CI: 9.3, 14.7). All responses were partial responses.
The most common adverse reactions (≥ 20%), excluding laboratory terms, were fatigue, bruising, cough, musculoskeletal pain, COVID-19, diarrhea, pneumonia, abdominal pain, dyspnea, hemorrhage, edema, nausea, pyrexia, and headache. Grade 3 or 4 laboratory abnormalities in greater than 10% of patients were decreased neutrophil counts, anemia, and decreased platelet counts. Serious infections occurred in 32% of patients including fatal infections in 10% of patients. The prescribing information includes warnings and precautions for infections, hemorrhage, cytopenias, cardiac arrhythmias, and secondary primary malignancies.
The recommended pirtobrutinib dose is 200 mg orally once daily until disease progression or unacceptable toxicity.
This review used the Assessment Aid, a voluntary submission from the applicant to facilitate the FDA’s assessment.
This application was granted priority review and orphan drug designation. FDA expedited programs are described in the Guidance for Industry: Expedited Programs for Serious Conditions-Drugs and Biologics.
Healthcare professionals should report all serious adverse events suspected to be associated with the use of any medicine and device to FDA’s MedWatch Reporting System or by calling 1-800-FDA-1088.
For assistance with single-patient INDs for investigational oncology products, healthcare professionals may contact OCE’s Project Facilitate at 240-402-0004 or email OncProjectFacilitate@fda.hhs.gov.

FDA approves repotrectinib for ROS1-positive non-small cell lung cancer
FDA approves repotrectinib for ROS1-positive non-small cell lung cancer
➤ First FDA approval that includes patients with ROS1-positive NSCLC who have previously received a ROS1 tyrosine kinase inhibitor (TKI), in addition to patients who are TKI naïve
➤ Traditional approval was granted on one single-arm study, utilizing high and durable responses for approval - 79% ORR in the ROS1 TKI naïve group, median DOR was 34.1 months; 38% ORR in those receiving prior treatment with a ROS1 inhibitor, median DOR 14.8 months

On November 15, 2023, the Food and Drug Administration approved repotrectinib (Augtyro, Bristol-Myers Squibb Company) for locally advanced or metastatic ROS1-positive non-small cell lung cancer (NSCLC).
This is the first FDA approval that includes patients with ROS1-positive NSCLC who have previously received a ROS1 tyrosine kinase inhibitor (TKI), in addition to patients who are TKI naïve.
The full prescribing information for Augtyro will be posted here.
Approval was based on TRIDENT-1, a global, multicenter, single-arm, open-label, multi-cohort clinical trial (NCT03093116) which included patients with ROS1-positive locally advanced or metastatic NSCLC. Efficacy was evaluated in 71 ROS1 TKI-naïve patients who received up to 1 prior line of platinum-based chemotherapy and/or immunotherapy and 56 patients who received 1 prior ROS1 TKI with no prior platinum-based chemotherapy or immunotherapy.
The major efficacy outcome measures were overall response rate (ORR) and duration of response (DOR) according to RECIST v1.1 as assessed by blinded independent central review. Confirmed ORR in the ROS1 TKI naïve group was 79% (95% CI: 68, 88) and 38% (95% CI: 25, 52) in those patients receiving prior treatment with a ROS1 inhibitor. Median DOR was 34.1 months (95% CI: 25.6, not evaluable) and 14.8 months (95% CI: 7.6, not evaluable) in the two respective groups. Responses were observed in intracranial lesions in patients with measurable CNS metastases, and in patients with resistance mutations following TKI therapy.
The most common (>20%) adverse reactions were dizziness, dysgeusia, peripheral neuropathy, constipation, dyspnea, ataxia, fatigue, cognitive disorders, and muscular weakness.
The recommended repotrectinib dose is 160 mg orally once daily with or without food for 14 days, then increased to 160 mg twice daily, until disease progression or unacceptable toxicity.
This review used the Assessment Aid, a voluntary submission from the applicant to facilitate the FDA’s assessment.
This application was granted priority review, breakthrough designation, and fast track designation. FDA expedited programs are described in the Guidance for Industry: Expedited Programs for Serious Conditions-Drugs and Biologics.

Truqap (capivasertib) plus Faslodex approved in the US for patients with advanced HR-positive breast cancer
Truqap (capivasertib) plus Faslodex approved in the US for patients with advanced HR-positive breast cancer
➤ First-in-class AKT inhibitor has potential to reshape treatment for breast cancer patients with specific biomarker alterations (PIK3CA, AKT1 or PTEN)
➤ Truqap+Faslodex reduced the risk of disease progression or death by 50% vs.Faslodex alone in patients with tumours harbouring PI3K/AKT pathway biomarker alterations (HR 0.50, 95% confidence interval 0.38-0.65; p=<0.001; median PFS 7.3 versus 3.1 months)

First-in-class AKT inhibitor has potential to reshape treatment for breast cancer patients with specific biomarker alterations (PIK3CA, AKT1 or PTEN)
Approval based on CAPItello-291 results which showed this combination reduced the risk of disease progression or death by 50% vs. Faslodex alone in the biomarker-altered population
AstraZeneca’s Truqap (capivasertib) in combination with Faslodex (fulvestrant) has been approved in the US for the treatment of adult patients with hormone receptor (HR)-positive, HER2-negative locally advanced or metastatic breast cancer with one or more biomarker alterations (PIK3CA, AKT1 or PTEN). Eligible patients will have progressed on at least one endocrine-based regimen in the metastatic setting or experienced recurrence on or within 12 months of completing adjuvant therapy.
The approval by the Food and Drug Administration (FDA) was based on the results from the CAPItello-291 Phase III trial published earlier this year in The New England Journal of Medicine.1 In the trial, Truqap in combination with Faslodex reduced the risk of disease progression or death by 50% versus Faslodex alone in patients with tumours harbouring PI3K/AKT pathway biomarker alterations (based on hazard ratio of 0.50, 95% confidence interval 0.38-0.65; p=<0.001; median progression-free survival (PFS) 7.3 versus 3.1 months).
Breast cancer is the most common cancer and one of the leading causes of cancer-related death worldwide.2 HR-positive breast cancer (expressing estrogen or progesterone receptors, or both), is the most common subtype, with more than 65% of tumours considered HR-positive and HER2-low or HER2-negative.3 Collectively, mutations in PIK3CA, AKT1 and alterations in PTEN occur frequently, affecting up to 50% of patients with advanced HR-positive breast cancer.4-6 Endocrine therapies are widely used in this setting, but many patients develop resistance to 1st-line cyclin-dependent kinase (CDK) 4/6 inhibitors and estrogen receptor-targeting therapies, underscoring the need for additional endocrine therapy-based options.7
Komal Jhaveri, MD, Medical Oncologist, Memorial Sloan Kettering Cancer Center (MSK), US, said: “Patients with advanced HR-positive breast cancer typically experience tumour progression or resistance with widely used first-line endocrine therapies and there is an urgent need to extend the effectiveness of these approaches. The combination of capivasertib and fulvestrant, a first-of-its-kind combination, provides a much-needed new treatment option for up to half of patients in this setting with these specific biomarkers, offering the potential to delay disease progression and provide more time with their disease under control.”
Dave Fredrickson, Executive Vice President, Oncology Business Unit, AstraZeneca, said: “The rapid US approval of Truqap reinforces the important role of the PI3K/AKT pathway in HR-positive breast cancer and the critical need to test patients at the time of diagnosis, as up to fifty per cent have tumours with these alterations. As a first-in-class medicine, this approval provides a critical new option for patients in the US with this specific type of disease and we look forward to bringing Truqap to the many breast cancer patients who can benefit across the globe.”
In the CAPItello-291 trial, the safety profile of Truqap plus Faslodex was similar to that observed in previous trials evaluating this combination.1
Concurrently with this approval, the FDA also approved a companion diagnostic test to detect relevant alterations (PIK3CA, AKT1 and PTEN).
The US regulatory submission was granted Priority Review and reviewed under Project Orbis, which provides a framework for concurrent submission and review of oncology medicines among participating international partners. As part of Project Orbis, Truqap plus Faslodex is also under review by regulatory authorities in Australia, Brazil, Canada, Israel, Singapore, Switzerland and the UK.
Regulatory applications for Truqap in combination with Faslodex are also currently under review in China, the European Union, Japan and several other countries.
Financial considerations
Following this approval in the US, Astex Therapeutics is eligible to receive a milestone payment from AstraZeneca on first commercial sale of the drug in the US as well as royalties on future sales in line with the agreement between the two companies.
Notes
HR-positive breast cancer
Breast cancer is the most common cancer and one of the leading causes of cancer-related death worldwide.2 More than two million patients were diagnosed with breast cancer in 2020, with nearly 685,000 deaths globally.2 In the US, more than 290,000 patients are expected to be diagnosed in 2023, with more than 43,000 deaths.8
HR-positive breast cancer (expressing estrogen or progesterone receptors, or both), is the most common subtype of breast cancer with more than 65% of tumours considered HR-positive and HER2-low or HER2-negative.3 Collectively, mutations in PIK3CA, AKT1 and alterations in PTEN occur frequently, affecting up to 50% of patients with advanced HR-positive breast cancer.4-6
The growth of HR-positive breast cancer cells is often driven by estrogen receptors (ER), and endocrine therapies that target ER-driven disease are widely used as first-line treatment in the advanced setting, and often paired with CDK4/6 inhibitors.7,9,10 However, resistance to CDK4/6 inhibitors and current endocrine therapies develops in many patients with advanced disease.9 Once this occurs, treatment options are limited – with chemotherapy being the current standard of care – and survival rates are low with 30% of patients anticipated to live beyond five years after diagnosis.3,9,11
The optimisation of endocrine therapy and overcoming resistance to enable patients to continue benefiting from these treatments, as well as identifying new therapies for those who are less likely to benefit, are active areas of focus for breast cancer research.
CAPItello-291
CAPItello-291 is a Phase III, double-blind, randomised trial evaluating the efficacy of Truqap in combination with Faslodex versus placebo plus Faslodex for the treatment of locally advanced (inoperable) or metastatic HR-positive, HER2-low or negative (immunohistochemistry (IHC) 0 or 1+, or IHC 2+/in-situ hybridisation (ISH)-negative) breast cancer.
The global trial enrolled 708 adult patients with histologically confirmed HR-positive, HER2-low or negative breast cancer whose disease has recurred or progressed during or after aromatase inhibitor therapy, with or without a CDK4/6 inhibitor, and up to one line of chemotherapy for advanced disease. The trial has dual primary endpoints of PFS in the overall patient population and in a population of patients whose tumours have qualifying alterations in the PI3K/AKT pathway (PIK3CA, AKT1 or PTEN genes). In the trial, approximately 40% of tumours had these alterations and approximately 70% of patients received a prior CDK4/6 inhibitor.
Truqap
Truqap (capivasertib) is a first-in-class, potent, adenosine triphosphate (ATP)-competitive inhibitor of all three AKT isoforms (AKT1/2/3). Truqap 400mg is administered twice daily according to an intermittent dosing schedule of four days on and three days off. This was chosen in early phase trials based on tolerability and the degree of target inhibition.
Truqap is currently being evaluated in Phase III trials for the treatment of multiple subtypes of breast cancer and in other tumour types either as monotherapy or in combination with established treatments. The ongoing clinical research programme is focused on tumours reliant on signalling via the PI3K/AKT pathway, and in tumours harbouring biomarker alterations in this pathway.
Truqap was discovered by AstraZeneca subsequent to a collaboration with Astex Therapeutics (and its collaboration with the Institute of Cancer Research and Cancer Research Technology Limited).
Faslodex
Faslodex is an endocrine therapy indicated for the treatment of estrogen receptor-positive, locally advanced or metastatic breast cancer in postmenopausal women not previously treated with endocrine therapy, or with disease relapse on or after adjuvant anti-estrogen therapy, or disease progression on anti-estrogen therapy.
In the US, EU and Japan, Faslodex is also approved in combination with CDK4/6 inhibitors for the treatment of women with HR-positive, HER2-negative advanced or metastatic breast cancer, whose cancer has progressed after endocrine medicine. Faslodex represents a hormonal treatment approach that helps to slow tumour growth by blocking and degrading the estrogen receptor – a key driver of disease progression.
Faslodex is approved as monotherapy or in combination with medicines from various drug classes including CDK4/6, PI3K and AKT inhibitors for the treatment of patients with HR-positive advanced breast cancer and is being evaluated in combination with medicines from other drug classes.
AstraZeneca in breast cancer
Driven by a growing understanding of breast cancer biology, AstraZeneca is starting to challenge, and redefine, the current clinical paradigm for how breast cancer is classified and treated to deliver even more effective treatments to patients in need – with the bold ambition to one day eliminate breast cancer as a cause of death.
AstraZeneca has a comprehensive portfolio of approved and promising compounds in development that leverage different mechanisms of action to address the biologically diverse breast cancer tumour environment.
With Enhertu (trastuzumab deruxtecan), a HER2-directed antibody drug conjugate (ADC), AstraZeneca and Daiichi Sankyo are aiming to improve outcomes in previously treated HER2-positive and HER2-low metastatic breast cancer and are exploring its potential in earlier lines of treatment and in new breast cancer settings.
In HR-positive breast cancer, AstraZeneca continues to improve outcomes with foundational medicines Faslodex and Zoladex (goserelin) and aims to reshape the HR-positive space with first-in-class AKT inhibitor, Truqap, and next-generation SERD and potential new medicine camizestrant. AstraZeneca is also collaborating with Daiichi Sankyo to explore the potential of TROP2-directed ADC, datopotamab deruxtecan, in this setting.
PARP inhibitor Lynparza (olaparib) is a targeted treatment option that has been studied in early and metastatic breast cancer patients with an inherited BRCA mutation. AstraZeneca with MSD (Merck & Co., Inc. in the US and Canada) continue to research Lynparza in these settings and to explore its potential in earlier disease.
To bring much-needed treatment options to patients with triple-negative breast cancer, an aggressive form of breast cancer, AstraZeneca is evaluating the potential of datopotamab deruxtecan alone and in combination with immunotherapy Imfinzi (durvalumab), Truqap in combination with chemotherapy, and Imfinzi in combination with other oncology medicines, including Lynparza and Enhertu.
AstraZeneca in oncology
AstraZeneca is leading a revolution in oncology with the ambition to provide cures for cancer in every form, following the science to understand cancer and all its complexities to discover, develop and deliver life-changing medicines to patients.
The Company's focus is on some of the most challenging cancers. It is through persistent innovation that AstraZeneca has built one of the most diverse portfolios and pipelines in the industry, with the potential to catalyse changes in the practice of medicine and transform the patient experience.
AstraZeneca has the vision to redefine cancer care and, one day, eliminate cancer as a cause of death.
AstraZeneca
AstraZeneca (LSE/STO/Nasdaq: AZN) is a global, science-led biopharmaceutical company that focuses on the discovery, development, and commercialisation of prescription medicines in Oncology, Rare Diseases, and BioPharmaceuticals, including Cardiovascular, Renal & Metabolism, and Respiratory & Immunology. Based in Cambridge, UK, AstraZeneca operates in over 100 countries and its innovative medicines are used by millions of patients worldwide. Please visit astrazeneca.com and follow the Company on Social Media @AstraZeneca.
Contacts
For details on how to contact the Investor Relations Team, please click here. For Media contacts, click here.
References
1. Turner N, et al. Capivasertib in Hormone Receptor–Positive Advanced Breast Cancer. NEJM. 2023; 388:2058–70.
2. Sung H, et al. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin. 2021; 10.3322/caac.21660.
3. National Cancer Institute. Surveillance, Epidemiology and End Results Program. Available at: https://seer.cancer.gov/statfacts/html/breast-subtypes.html. Accessed November 2023.
4. Howell S J, et al. Fulvestrant plus capivasertib versus placebo after relapse or progression on an aromatase inhibitor in metastatic, oestrogen receptor-positive, HER2-negative breast cancer (FAKTION). J Clin Oncol. 2022; 23:851-64.
5. Hortobagyi G N, et al. Correlative Analysis of Genetic Alterations and Everolimus Benefit in Hormone Receptor-Positive, Human Epidermal Growth Factor Receptor 2-Negative Advanced Breast Cancer: Results From BOLERO-2. J Clin Oncol. 2016; 34:419-26.
6. Millis S Z, et al. Landscape of phosphatidylinositol-3-kinase pathway alterations across 19784 diverse solid tumors. JAMA Oncol. 2016;2(12):1565-73.
7. Lin M, et al. Comparative Overall Survival of CDK4/6 Inhibitors Plus Endocrine Therapy vs. Endocrine Therapy Alone for Hormone receptor-positive, HER2-negative metastatic breast cancer. J Cancer. 2020; 10.7150/jca.48944.
8. American Cancer Society. Key Statistics for Breast Cancer. Available at: https://www.cancer.org/cancer/breast-cancer/about/how-common-is-breast-cancer.html. Accessed November 2023.
9. Lloyd M R, et al. Mechanisms of Resistance to CDK4/6 Blockade in Advanced Hormone Receptor–positive, HER2-negative Breast Cancer and Emerging Therapeutic Opportunities. Clin Cancer Res. 2022; 28(5):821-30.
10. Scabia V, et al. Estrogen receptor positive breast cancers have patient specific hormone sensitivities and rely on progesterone receptor. Nat Commun. 2022; 10.1038/s41467-022-30898-0.
11. National Comprehensive Cancer Network. Clinical Practice Guidelines in Oncology (NCCN Guidelines). Available at: https://www.nccn.org/guidelines/guidelines-detail?category=1&id=1419. Accessed November 2023.
Dr. Jhaveri has financial interests related to AstraZeneca.

BeiGene Receives European Commission Approval for BRUKINSA® (zanubrutinib) for the Treatment of Relapsed or Refractory Follicular Lymphoma
BeiGene Receives European Commission Approval for BRUKINSA® (zanubrutinib) for the Treatment of Relapsed or Refractory Follicular Lymphoma
BRUKINSA is the first and only BTK inhibitor approved for follicular lymphoma in the European Union
ORR was 69.0% in the BRUKINSA plus obinutuzumab arm versus 45.8% in the obinutuzumab arm (P = 0.0012), with a median follow-up of approximately 20 months
Responses were durable with an 18-month DOR of 69.3% in the BRUKINSA combination arm
Median PFS was 28.0 months vs. 10.4 months for obinutuzumab alone (HR: 0.50 [95% CI: 0.33, 0.75]; P = 0.0007)

BRUKINSA is the first and only BTK inhibitor approved for follicular lymphoma in the European Union
Approval was based on results from the ROSEWOOD trial in which BRUKINSA plus the anti-CD20 monoclonal antibody obinutuzumab achieved higher overall response rate compared to obinutuzumab alone
BASEL, Switzerland & BEIJING & CAMBRIDGE, Mass.--(BUSINESS WIRE)-- BeiGene, Ltd. (Nasdaq: BGNE; HKEX: 06160; SSE: 688235), a global biotechnology company, today announced that the European Commission (EC) has granted marketing authorization for BRUKINSA® (zanubrutinib) in combination with obinutuzumab for the treatment of adult patients with relapsed or refractory (R/R) follicular lymphoma (FL) who have received at least two prior lines of systemic therapy. This marks the fourth indication in the European Union (EU) for BRUKINSA, which is now approved to treat more patient populations in the EU than any other Bruton’s tyrosine kinase (BTK) inhibitor.
“With this approval, we are excited to announce that BRUKINSA will become available as a treatment option for patients with follicular lymphoma in the European Union. BRUKINSA is now the first BTK inhibitor approved in this indication and has the broadest label of any medicine in its class globally,” said Mehrdad Mobasher, M.D., M.P.H., Chief Medical Officer, Hematology at BeiGene. “This milestone marks a significant advancement in our efforts to combat the disease by providing a new and effective treatment option to patients who have either failed to respond to initial therapies or have experienced a relapse.”
The EC approval is based on positive results from ROSEWOOD (NCT03332017), a global, randomized, open-label Phase 2 study of BRUKINSA plus obinutuzumab compared with obinutuzumab alone in 217 patients with R/R FL who received at least two prior lines of systemic therapy. In the study, the overall response rate was 69.0% in the BRUKINSA plus obinutuzumab arm versus 45.8% in the obinutuzumab arm (P = 0.0012), with a median follow-up of approximately 20 months. Responses were durable with 18-month landmark duration of response (DOR) of 69.3% in the BRUKINSA combination arm.
Additionally, the median progression-free survival (PFS) for patients treated with BRUKINSA plus obinutuzumab was 28.0 months, compared to 10.4 months for patients treated with only obinutuzumab (HR: 0.50 [95% CI: 0.33, 0.75]; P = 0.0007).
BRUKINSA plus obinutuzumab was generally well-tolerated, with safety results consistent with previous studies of both medicines.
“People living with follicular lymphoma often experience relapse and have poor responses to subsequent lines of therapy, making it imperative to improve outcomes,” said Pier Luigi Zinzani, M.D., Ph.D., Full Professor of Haematology at the Institute of Haematology “Seràgnoli,” University of Bologna, Italy. “The results from the ROSEWOOD trial demonstrated a significant clinical benefit of BRUKINSA plus obinutuzumab for patients with relapsed or refractory follicular lymphoma. BRUKINSA is a chemotherapy-free, oral treatment option that can be a practice-changing option for eligible patients with relapsed or refractory follicular lymphoma.”
In addition to R/R FL, BRUKINSA is approved in the EU as monotherapy for the treatment of adult patients with chronic lymphocytic leukemia, for adult patients with marginal zone lymphoma who have received at least one prior anti-CD20-based therapy, and for adult patients with Waldenström’s macroglobulinemia who have received at least one prior therapy, or in first-line treatment for patients unsuitable for chemo-immunotherapy.
Gerwin Winter, Senior Vice President, Head of Europe at BeiGene noted, “We have made great progress in making BRUKINSA available to eligible patients with hematological malignancies globally, and this approval is a testament to our continued commitment to bring this much needed treatment option to patients in Europe and around the world. We hope that this approval will have a positive impact on the lives of many people living with follicular lymphoma in the European Union and their families.”
BeiGene currently has submissions for BRUKINSA in R/R FL under review by regulatory authorities including in the United States (U.S.) and China. Additionally, BeiGene’s submission for BRUKINSA in R/R FL is under review by regulatory authorities in Canada, Switzerland, and the United Kingdom as part of the Access Consortium New Active Substance Work-sharing Initiative (NASWSI).
BRUKINSA is approved in more than 65 markets, including the U.S., China, EU, Great Britain, Canada, Australia, South Korea, and Switzerland in selected indications and under development for additional indications globally. Product information may differ from country to country. Prescribers should consult the product information approved in their respective countries. The global BRUKINSA development program includes more than 5,000 subjects enrolled to date in 29 countries and regions.
The Summary of Product Characteristics for BRUKINSA can be found here: https://www.ema.europa.eu/en/documents/product-information/brukinsa-epar-product-information_en.pdf
About Follicular Lymphoma
FL is the second most common type of non-Hodgkin lymphoma (NHL), accounting for 22 percent of all NHL cases.i Across Europe, over 122,000 people each year are diagnosed with NHL.ii FL is a slow-growing cancer but can become more aggressive over time. While FL remains incurable, people with the condition can live a long time. The five-year survival rate is about 90 percent, and approximately half of people diagnosed with FL can live with the disease for nearly 20 years.iii,iv
About BRUKINSA® (zanubrutinib)
BRUKINSA is a small molecule inhibitor of Bruton’s tyrosine kinase (BTK) discovered by BeiGene scientists that is currently being evaluated globally in a broad clinical program as a monotherapy and in combination with other therapies to treat various B-cell malignancies. Because new BTK is continuously synthesized, BRUKINSA was specifically designed to deliver complete and sustained inhibition of the BTK protein by optimizing bioavailability, half-life, and selectivity. With differentiated pharmacokinetics compared to other approved BTK inhibitors, BRUKINSA has been demonstrated to inhibit the proliferation of malignant B cells within a number of disease relevant tissues.
About BeiGene
BeiGene is a global biotechnology company that is discovering and developing innovative oncology treatments that are more affordable and accessible to cancer patients worldwide. With a broad portfolio, we are expediting development of our diverse pipeline of novel therapeutics through our internal capabilities and collaborations. We are committed to radically improving access to medicines for far more patients who need them. Our growing global team of more than 10,000 colleagues spans five continents, with administrative offices in Basel, Beijing, and Cambridge, U.S. To learn more about BeiGene, please visit www.beigene.com and follow us on LinkedIn and X (formerly known as Twitter).
Forward-Looking Statements
This press release contains forward-looking statements within the meaning of the Private Securities Litigation Reform Act of 1995 and other federal securities laws, including statements regarding BeiGene’s ability to provide effective treatment options to patients with FL; whether BRUKINSA is a practice-changing option for eligible patients; the effect, if any, that the EC approval of BRUKINSA for R/R FL will have on people living with R/R FL and their families; BeiGene’s advancement, anticipated clinical development, regulatory submissions and commercialization of zanubrutinib, particularly as a treatment for R/R FL; and BeiGene’s plans, commitments, aspirations, and goals under the heading “About BeiGene.” Actual results may differ materially from those indicated in the forward-looking statements as a result of various important factors, including BeiGene's ability to demonstrate the efficacy and safety of its drug candidates; the clinical results for its drug candidates, which may not support further development or marketing approval; actions of regulatory agencies, which may affect the initiation, timing and progress of clinical trials and marketing approval; BeiGene's ability to achieve commercial success for its marketed medicines and drug candidates, if approved; BeiGene's ability to obtain and maintain protection of intellectual property for its medicines and technology; BeiGene's reliance on third parties to conduct drug development, manufacturing, commercialization, and other services; BeiGene’s limited experience in obtaining regulatory approvals and commercializing pharmaceutical products and its ability to obtain additional funding for operations and to complete the development of its drug candidates and achieve and maintain profitability; and those risks more fully discussed in the section entitled “Risk Factors” in BeiGene’s most recent quarterly report on Form 10-Q, as well as discussions of potential risks, uncertainties, and other important factors in BeiGene's subsequent filings with the U.S. Securities and Exchange Commission. All information in this press release is as of the date of this press release, and BeiGene undertakes no duty to update such information unless required by law.
References
iLeukemia & Lymphoma Society. Treatment for Indolent NHL Subtypes. Available at: https://www.lls.org/lymphoma/non-hodgkin-lymphoma/nhl-subtypes/treatment-indolent-nhl-subtypes.
iiWorld Health Organization. Non-Hodgkin Lymphoma. Available at: https://gco.iarc.fr/today/data/factsheets/cancers/34-Non-hodgkin-lymphoma-fact-sheet.pdf.
iiiAmerican Cancer Society. Survival Rates and Factors That Affect Prognosis (Outlook) for Non-Hodgkin Lymphoma. Available at: https://www.cancer.org/cancer/types/non-hodgkin-lymphoma/detection-diagnosis-staging/factors-prognosis.
ivCartron G and Trotman J. Time for an Individualized Approach to First-Line Management of Follicular Lymphoma. Haematologica. 2022;107(1):7-18.

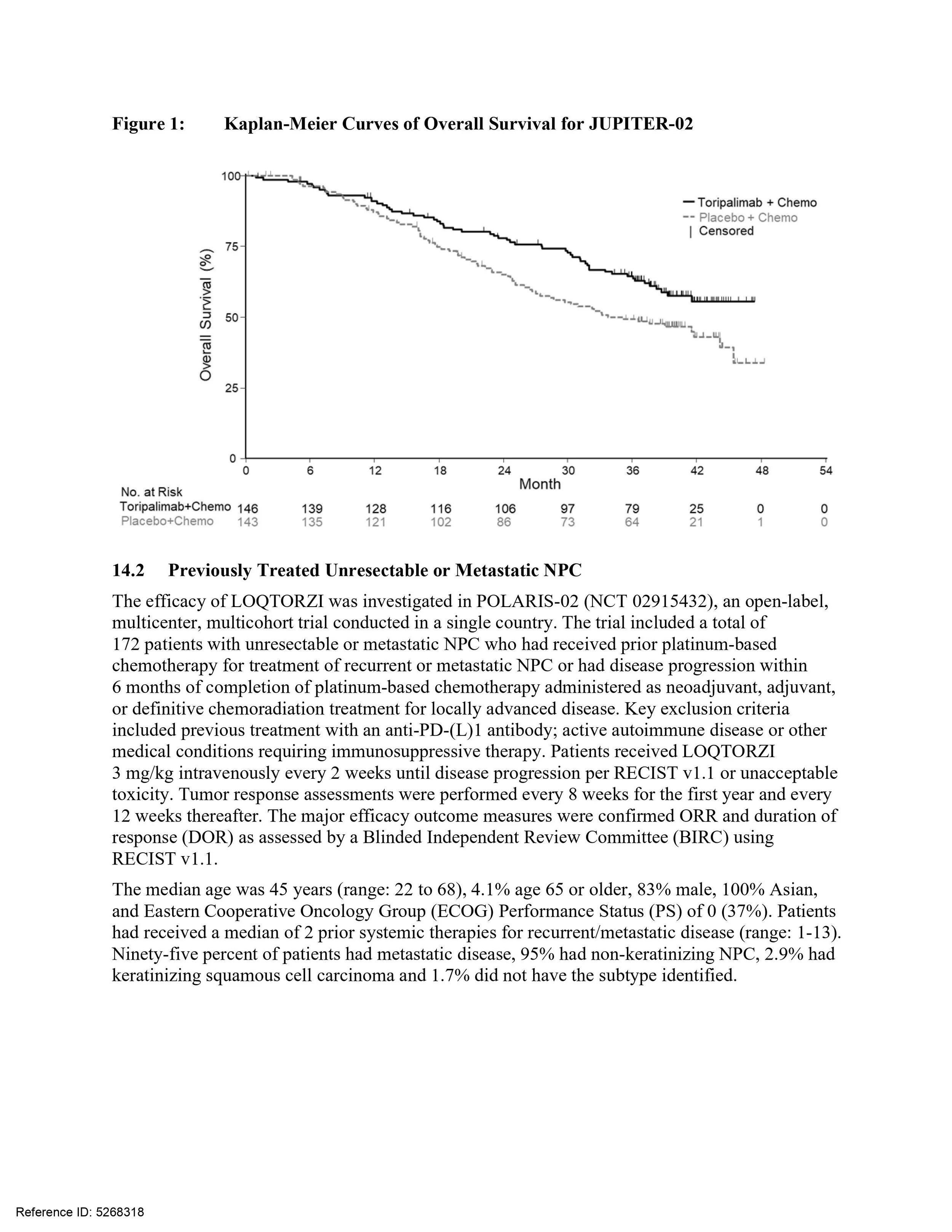
FDA approves first novel PD-1 developed in China; US trial to come post-approval
FDA approves first novel PD-1 developed in China; US trial to come post-approval
100% of pivotal study participants were Asian. Approval comes with a post-marketing commitment to conduct a clinical trial enrolling 100 patients in the U.S. and Canada, that includes a sufficient representation of patients in racial and ethnic minority subgroups and is reflective of the U.S. population of patients with nasopharyngeal carcinoma (NPC)
Loqtorzi (toripalimab) is the first FDA approved treatment for nasopharyngeal carcinoma
Developed by Shanghai Junshi Biosciences in collaboration with Coherus BioSciences, Loqtorzi becomes the first Chinese PD-1 to gain FDA approval



























European approval for AbbVie's TEPKINLY® (epcoritamab) for Adults with Relapsed or Refractory Diffuse Large B-cell Lymphoma
European approval for AbbVie's TEPKINLY® (epcoritamab) for Adults with Relapsed or Refractory Diffuse Large B-cell Lymphoma
TEPKINLY® (epcoritamab) is the first and only subcutaneous bispecific antibody approved as a monotherapy for adult patients with relapsed or refractory diffuse large B-cell lymphoma after two or more lines of systemic therapy
Conditional marketing authorization approval from the European Commission is supported by data from the pivotal Phase 1/2 EPCORE™ NHL-1 clinical trial
ORR 62% & a CR rate of 39%; median DOR was 15.5 months (range: 9.7, not reached)

TEPKINLY® (epcoritamab) is the first and only subcutaneous bispecific antibody approved as a monotherapy for adult patients with relapsed or refractory diffuse large B-cell lymphoma after two or more lines of systemic therapy
Conditional marketing authorization approval from the European Commission is supported by data from the pivotal Phase 1/2 EPCORE™ NHL-1 clinical trial
TEPKINLY represents AbbVie's second approved hematological cancer treatment in the European Union
NORTH CHICAGO, Ill., Sept. 25, 2023 /PRNewswire/ -- AbbVie (NYSE: ABBV) today announced that the European Commission (EC) has granted conditional marketing authorization for TEPKINLY® (epcoritamab) as a monotherapy for the treatment of adult patients with relapsed or refractory (R/R) diffuse large B-cell lymphoma (DLBCL) after two or more lines of systemic therapy. TEPKINLY is the first and only subcutaneous T-cell engaging bispecific antibody approved for the treatment of this patient population in the European Union (EU), as well as Liechtenstein, Norway and Iceland.
DLBCL is the most common type of B-cell non-Hodgkin's lymphoma worldwide.1 While patients may have access to chemoimmunotherapy regimens to treat their disease, they face limited treatment options, with few readily available, off-the-shelf medicines, especially for those whose disease has relapsed or become refractory to prior treatments.1
"The European Commission approval of epcoritamab represents a significant milestone in our aspiration with Genmab to develop a potential core therapy for patients with B-cell malignancies, like DLBCL," said Roopal Thakkar, senior vice president, development and regulatory affairs, chief medical officer, AbbVie. "With this milestone achievement, TEPKINLY is now the second approved cancer treatment in the EU from our oncology portfolio, and AbbVie's third blood cancer medicine across the world. We remain committed to developing new innovative medicines that help improve the lives of people with hematological cancers."
This conditional approval is supported by data from the pivotal EPCORE™ NHL-1 Phase 1/2 open-label, multi-cohort, multi-center, single-arm trial evaluating the preliminary efficacy and safety of TEPKINLY in patients with R/R large B-cell lymphoma (LBCL), including its subtype DLBCL. In this study, DLBCL patients treated with TEPKINLY (N=139) achieved an overall response rate of 62 percent and a complete response rate of 39 percent. The median duration of response was 15.5 months (range: 9.7, not reached).
Results from the trial showed that TEPKINLY demonstrated a manageable safety profile across the LBCL patient cohort (N=167), which included the DLBCL patient population. The most common adverse reactions (≥ 20 percent) were cytokine release syndrome, fatigue, neutropenia, injection site reaction, musculoskeletal pain, abdominal pain, pyrexia, nausea and diarrhea.
"Relapsed or refractory DLBCL is an aggressive cancer and patients can face a difficult and emotional treatment journey. At this point in the journey, a patient may have had multiple lines of therapy and will already have experienced relapse," said Anna Sureda, M.D., Ph.D., head of clinical hematology department, Institut Català d'Oncologia – L'Hospitalet, Barcelona, Spain. "This European Commission approval represents an important moment for the DLBCL patient community and brings with it a potential opportunity for effective disease management for a condition with limited available treatment options."
Conditional marketing authorization is granted to medicines that address an unmet medical need, where the benefit of its immediate availability to patients outweighs the risk of limited data availability, and where comprehensive data will be provided.2
TEPKINLY is being co-developed by AbbVie and Genmab as part of the companies' oncology collaboration. The companies will share commercial responsibilities in the U.S. and Japan, with AbbVie responsible for further global commercialization. AbbVie will continue to pursue regulatory submissions for epcoritamab across international markets throughout the year.
About the EPCORE™ NHL-1 Trial
EPCORE™ NHL-1 evaluated epcoritamab as a monotherapy in patients with CD20+ relapsed or refractory (R/R) large B-cell lymphoma (LBCL), including diffuse large B-cell lymphoma (DLBCL), after two or more lines of systemic therapy.3 The study included a dose escalation part and an expansion part.3 The primary efficacy endpoint was overall response rate determined by Lugano criteria (2014) as assessed by an independent review committee.3 More information can be found on www.clinicaltrials.gov.
About TEPKINLY® (epcoritamab)
TEPKINLY is an investigational IgG1-bispecific antibody created using Genmab's proprietary DuoBody® technology. Genmab's DuoBody®-CD3 technology is designed to direct cytotoxic T-cells selectively to elicit an immune response toward target cell types. TEPKINLY is designed to simultaneously bind to CD3 on T-cells and CD20 on B-cells and induces T-cell mediated killing of CD20+ cells.4 CD20 is expressed on B-cells and is a clinically validated therapeutic target in many B-cell malignancies, including DLBCL, follicular lymphoma, mantle cell lymphoma and chronic lymphocytic leukemia.5,6
The U.S. Food and Drug Administration (FDA) approved epcoritamab under the brand name EPKINLY™ (epcoritamab-bysp) in May 2023 for the treatment of adult patients with R/R DLBCL, not otherwise specified, including DLBCL arising from indolent lymphoma, and high-grade B-cell lymphoma, after two or more lines of systemic therapy. EPKINLY is approved under the FDA's Accelerated Approval program based on response rate and durability of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in confirmatory trials.
EU Indications and Important Safety Information about Tepkinly® ▼ (epcoritamab)
Indications
Tepkinly (epcoritamab) as monotherapy is indicated for the treatment of adult patients with relapsed or refractory diffuse large B-cell lymphoma (DLBCL) after two or more lines of systemic therapy.
Important Safety Information
Contraindications
Hypersensitivity to the active substance or to any of the excipients.
Special warnings and precautions for use
Cytokine release syndrome (CRS)
CRS, which may be life-threatening or fatal, occurred in patients receiving Tepkinly. The most common signs and symptoms of CRS include pyrexia, hypotension and hypoxia. Other signs and symptoms of CRS in more than two patients include chills, tachycardia, headache and dyspnoea.
Most CRS events occurred in Cycle 1 and were associated with the first full dose of Tepkinly. Administer prophylactic corticosteroids to mitigate the risk of CRS. Patients should be monitored for signs and symptoms of CRS following Tepkinly administration. Patients should be hospitalised for 24 hours after administration of the Cycle 1 Day 15 dose of 48 mg to monitor for signs and symptoms of CRS. At the first signs or symptoms of CRS, institute treatment of supportive care with tocilizumab and/or corticosteroids as appropriate. Patients should be counselled on the signs and symptoms associated with CRS and patients should be instructed to contact their healthcare professional and seek immediate medical attention should signs or symptoms occur at any time. Management of CRS may require either temporary delay or discontinuation of Tepkinly based on the severity of CRS.
Immune effector cell-associated neurotoxicity syndrome (ICANS)
ICANS, including a fatal event, have occurred in patients receiving Tepkinly. ICANS may manifest as aphasia, altered level of consciousness, impairment of cognitive skills, motor weakness, seizures, and cerebral oedema. The majority of cases of ICANS occurred within Cycle 1 of Tepkinly treatment, however some occurred with delayed onset. Patients should be monitored for signs and symptoms of ICANS following Tepkinly administration. Patients should be hospitalised for 24 hours after administration of the Cycle 1 Day 15 dose of 48 mg to monitor for signs and symptoms of ICANS. At the first signs or symptoms of ICANS treatment with corticosteroids and non-sedating-anti-seizure medicinal products should be instituted as appropriate. Patients should be counselled on the signs and symptoms of ICANS and that the onset of events may be delayed. Patients should be instructed to contact their healthcare professional and seek immediate medical attention should signs or symptoms occur at any time. Tepkinly should be delayed or discontinued as recommended.
Serious infections
Treatment with Tepkinly may lead to an increased risk of infections. Serious or fatal infections were observed in patients treated with Tepkinly in clinical studies. Administration of Tepkinly should be avoided in patients with clinically significant active systemic infections. As appropriate, prophylactic antimicrobials should be administered prior to and during treatment with Tepkinly. Patients should be monitored for signs and symptoms of infection, before and after Tepkinly administration, and treated appropriately. In the event of febrile neutropenia, patients should be evaluated for infection and managed with antibiotics, fluids and other supportive care, according to local guidelines.
Tumour Lysis Syndrome (TLS)
TLS has been reported in patients receiving Tepkinly. Patients at an increased risk for TLS are recommended to receive hydration and prophylactic treatment with a uric acid lowering agent. Patients should be monitored for signs or symptoms of TLS, especially patients with high tumour burden or rapidly proliferative tumours, and patients with reduced renal function. Patients should be monitored for blood chemistries and abnormalities should be managed promptly.
Tumour flare
Tumour flare has been reported in patients treated with Tepkinly. Manifestations could include localized pain and swelling. Consistent with the mechanism of action of Tepkinly, tumour flare is likely due to the influx of T-cells into tumour sites following Tepkinly administration. There are no specific risk factors for tumour flare that have been identified; however, there is a heightened risk of compromise and morbidity due to mass effect secondary to tumour flare in patients with bulky tumours located in close proximity to airways and/or a vital organ. Patients treated with Tepkinly should be monitored and evaluated for tumour flare at critical anatomical sites.
CD20-negative disease
There are limited data available on patients with CD20-negative DLBCL treated with Tepkinly, and it is possible that patients with CD20-negative DLBCL may have less benefit compared to patients with CD20-positive DLBCL. The potential risks and benefits associated with treatment of patients with CD20-negative DLBCL with Tepkinly should be considered.
Immunisation
Live and/or live-attenuated vaccines should not be given during Tepkinly therapy. Studies have not been conducted in patients who received live vaccines.
Fertility, pregnancy and lactation
Tepkinly is not recommended during pregnancy and in women of childbearing potential not using contraception.
Effects on ability to drive and use machines
Tepkinly has minor influence on the ability to drive and use machines. Due to the potential for ICANS, patients should be advised to exercise caution while (or avoid if symptomatic) driving, cycling or using heavy or potentially dangerous machines.
Undesirable effects
Summary of the safety profile
The most common adverse reactions (≥ 20%) were CRS, fatigue, neutropenia, injection site reactions, musculoskeletal pain, abdominal pain, pyrexia, nausea, and diarrhoea. Serious adverse reactions occurred in 52% of patients. The most frequent serious adverse reaction (≥ 10%) was cytokine release syndrome (31%). Seven patients (4.2%) experienced a fatal adverse reaction (pneumonia in 3 (1.8%) patients, viral infection in 3 (1.8%) patients and ICANS in 1 (0.6%) patient). Adverse reactions that led to discontinuation occurred in 6.6% of patients. Discontinuation of Tepkinly due to pneumonia occurred in 6 (3.6%) patients, viral infection in 3 (1.8%) patients, and CRS, ICANS, or fatigue in 1 (0.6%) patient each. Dose delays due to adverse reactions occurred in 32% of patients. Adverse reactions leading to dose delays (≥ 3%) were viral infections (9.6%), CRS (7.2%), neutropenia (4.8%), pyrexia (3.0%), and thrombocytopenia (3.0%).
This is not a complete summary of all safety information.
See Tepkinly® full Summary of Product Characteristics (SmPC) at www.ema.europa.eu
Globally, prescribing information varies; refer to the individual country product label for complete information.
EPKINLY™ (epcoritamab-bysp) U.S. IMPORTANT SAFETY INFORMATION
Important Warnings—EPKINLY can cause serious side effects, including:
Cytokine Release Syndrome (CRS). CRS is common during treatment with EPKINLY and can be serious or life-threatening. Tell your healthcare provider or get medical help right away if you develop symptoms of CRS, including fever of 100.4°F (38°C) or higher, dizziness or lightheadedness, trouble breathing, chills, fast heartbeat, feeling anxious, headache, confusion, shaking (tremors), or problems with balance and movement, such as trouble walking.
Due to the risk of CRS, you will receive EPKINLY on a "step-up" dosing schedule. The step-up dosing schedule is when you receive smaller "step-up" doses of EPKINLY on day 1 and day 8 of your first cycle of treatment (cycle 1). You will receive your first full dose of EPKINLY on day 15 of cycle 1. If your dose of EPKINLY is delayed for any reason, you may need to repeat the step-up dosing schedule. Before each dose in cycle 1, you will receive medicines to help reduce your risk of CRS. Your healthcare provider will decide if you need to receive medicine to help reduce your risk of CRS with future cycles.
Neurologic problems. EPKINLY can cause serious neurologic problems that can be life-threatening and lead to death. Neurologic problems may happen days or weeks after you receive EPKINLY. Your healthcare provider may refer you to a healthcare provider who specializes in neurologic problems. Tell your healthcare provider right away if you develop any symptoms of neurologic problems, including trouble speaking or writing, confusion and disorientation, drowsiness, tiredness or lack of energy, muscle weakness, shaking (tremors), seizures, or memory loss.
Due to the risk of CRS and neurologic problems, you should be hospitalized for 24 hours after receiving your first full dose of EPKINLY on day 15 of cycle 1. Your healthcare provider will monitor you for symptoms of CRS and neurologic problems during treatment with EPKINLY, as well as other side effects, and treat you if needed. Your healthcare provider may temporarily stop or completely stop your treatment with EPKINLY if you develop CRS, neurologic problems, or any other side effects that are severe.
Do not drive or use heavy or potentially dangerous machinery if you develop dizziness, confusion, tremors, drowsiness, or any other symptoms that impair consciousness until your symptoms go away. These may be symptoms of CRS or neurologic problems.
EPKINLY can also cause other serious side effects, including:
Infections. EPKINLY can cause serious infections that may lead to death. Your healthcare provider will check you for symptoms of infection before and during treatment. Tell your healthcare provider right away if you develop any symptoms of infection during treatment, including fever of 100.4°F (38°C) or higher, cough, chest pain, tiredness, shortness of breath, painful rash, sore throat, pain during urination, or feeling weak or generally unwell.
Low blood cell counts. Low blood cell counts are common during treatment with EPKINLY and can be serious or severe. Your healthcare provider will check your blood cell counts during treatment. EPKINLY may cause low blood cell counts, including low white blood cell counts (neutropenia), which can increase your risk for infection; low red blood cell counts (anemia), which can cause tiredness and shortness of breath; and low platelet counts (thrombocytopenia), which can cause bruising or bleeding problems.
Your healthcare provider may temporarily stop or completely stop treatment with EPKINLY if you develop certain side effects.
Before you receive EPKINLY, tell your healthcare provider about all of your medical conditions, including if you:
have an infection.
are pregnant or plan to become pregnant. EPKINLY may harm your unborn baby. Females who are able to become pregnant: Your healthcare provider should do a pregnancy test before you start treatment with EPKINLY. You should use effective birth control (contraception) during treatment and for 4 months after your last dose of EPKINLY. Tell your healthcare provider if you become pregnant or think that you may be pregnant during treatment with EPKINLY.
are breastfeeding or plan to breastfeed. It is not known if EPKINLY passes into your breast milk. Do not breastfeed during treatment with EPKINLY and for 4 months after your last dose of EPKINLY.
Tell your healthcare provider about all of the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements.
The most common side effects of EPKINLY include CRS, tiredness, muscle and bone pain, injection site reactions, fever, stomach-area (abdominal) pain, nausea, and diarrhea.
These are not all the possible side effects of EPKINLY. Call your doctor for medical advice about side effects.
You are encouraged to report side effects to the FDA at (800) FDA-1088 or www.fda.gov/medwatch or to Genmab US, Inc. at 1-855-4GENMAB (1-855-443-6622).
Please see the Full Prescribing Information and Medication Guide, including Important Warnings.
About AbbVie in Oncology
At AbbVie, we are committed to transforming standards of care for multiple blood cancers while advancing a dynamic pipeline of investigational therapies across a range of cancer types. Our dedicated and experienced team joins forces with innovative partners to accelerate the delivery of potential breakthrough medicines. We are evaluating more than 20 investigational medicines in over 300 clinical trials across some of the world's most widespread and debilitating cancers. As we work to have a remarkable impact on people's lives, we are committed to exploring solutions to help patients obtain access to our cancer medicines. For more information, please visit http://www.abbvie.com/oncology.
About AbbVie
AbbVie's mission is to discover and deliver innovative medicines and solutions that solve serious health issues today and address the medical challenges of tomorrow. We strive to have a remarkable impact on people's lives across several key therapeutic areas – immunology, oncology, neuroscience, and eye care – and products and services in our Allergan Aesthetics portfolio. For more information about AbbVie, please visit us at www.abbvie.com. Follow @abbvie on LinkedIn, Facebook, Instagram, X (formerly Twitter), and YouTube.
AbbVie Forward-Looking Statements
Some statements in this news release are, or may be considered, forward-looking statements for purposes of the Private Securities Litigation Reform Act of 1995. The words "believe," "expect," "anticipate," "project" and similar expressions and uses of future or conditional verbs, generally identify forward-looking statements. AbbVie cautions that these forward-looking statements are subject to risks and uncertainties that may cause actual results to differ materially from those expressed or implied in the forward-looking statements. Such risks and uncertainties include, but are not limited to, challenges to intellectual property, competition from other products, difficulties inherent in the research and development process, adverse litigation or government action, and changes to laws and regulations applicable to our industry. Additional information about the economic, competitive, governmental, technological and other factors that may affect AbbVie's operations is set forth in Item 1A, "Risk Factors," of AbbVie's 2022 Annual Report on Form 10-K, which has been filed with the Securities and Exchange Commission, as updated by its subsequent Quarterly Reports on Form 10-Q. AbbVie undertakes no obligation, and specifically declines, to release publicly any revisions to forward-looking statements as a result of subsequent events or developments, except as required by law.
1 Sehn, Salles. "Diffuse Large B-Cell Lymphoma." N Engl J Med. 2021;384:842-858. DOI: 10.1056/NEJMra2027612.
2 European Medicines Agency. Conditional Marketing Authorisation. http://www.ema.europa.eu/ema/index.jsp?curl=pages/regulation/general/general_content_000925.jsp. Accessed August 2023.
3 First-in-human (FIH) trial in patients with relapsed, progressive or refractory B-cell lymphoma - clinicaltrials.gov. in. (n.d.). https://classic.clinicaltrials.gov/ct2/show/NCT03625037. Accessed August 2023.
4 Engelberts et al. "DuoBody-CD3xCD20 induces potent T-cell-mediated killing of malignant B cells in preclinical models and provides opportunities for subcutaneous dosing." EBioMedicine. 2020;52:102625. DOI: 10.1016/j.ebiom.2019.102625.
5 Rafiq, Butchar, Cheney, et al. "Comparative Assessment of Clinically Utilized CD20-Directed Antibodies in Chronic Lymphocytic Leukemia Cells Reveals Divergent NK Cell, Monocyte, and Macrophage Properties." J. Immunol. 2013;190(6):2702-2711. DOI: 10.4049/jimmunol.1202588.
6 Singh, Gupta, Almasan. "Development of Novel Anti-Cd20 Monoclonal Antibodies and Modulation in Cd20 Levels on Cell Surface: Looking to Improve Immunotherapy Response." J Cancer Sci Ther. 2015;7(11):347-358. DOI: 10.4172/1948-5956.1000373.